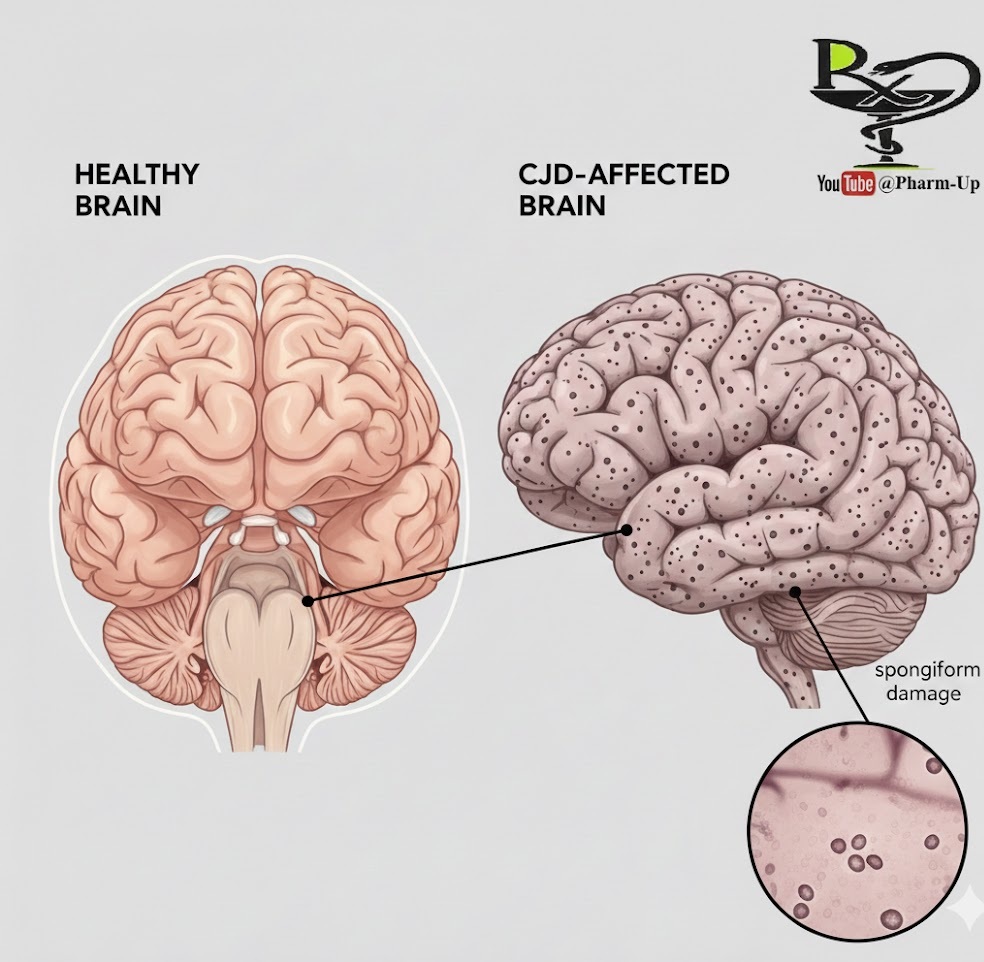
Creutzfeldt-Jakob disease (CJD) is a rare and aggressive degenerative brain disorder. It belongs to a family of human and animal diseases known as transmissible spongiform encephalopathies (TSEs). The disease is characterized by a rapid decline in mental health and physical coordination, typically leading to death within a year of symptom onset.
The Biological Mechanism: Prions
CJD is unique because it is caused by prions—misfolded proteins that lack DNA or RNA. When these proteins misfold, they trigger a chain reaction that causes healthy proteins in the brain to misfold as well. This leads to:
- Neuronal Death: The buildup of these proteins is toxic to brain cells.
- Brain Cavitation: Under a microscope, the brain develops tiny holes, resembling a sponge (spongiosis).
Classification of CJD
There are three primary ways the disease manifests:
- Sporadic CJD: The most common form (85% of cases). It occurs spontaneously when normal proteins randomly begin to misfold.
- Hereditary (Familial) CJD: Caused by a mutation in the PRNP gene that provides instructions for making prion proteins. This is passed down through families.
- Acquired CJD: The rarest form, contracted through exposure to infected brain or nervous system tissue. This historically occurred through contaminated surgical instruments or certain medical treatments.
The “Mad Cow” Connection (vCJD)
Variant CJD (vCJD) is a specific type linked to the consumption of beef infected with Bovine Spongiform Encephalopathy (BSE). While classic CJD usually affects seniors around age 60, vCJD often affects much younger patients and has a slightly slower progression.
Symptoms and Progression
Because the damage is so widespread, symptoms appear across multiple systems:
- Cognitive: Rapidly progressing dementia and memory loss.
- Physical: Involuntary muscle jerks (myoclonus), loss of balance, and slurred speech.
- Sensory: Blurred vision or blindness.
- Terminal Stage: Patients eventually lose the ability to speak or move, entering a coma before passing.